


Abstract
Pancreatic cancer is characterized by aberrant activity of oncogenic KRAS, which is mutated in 90% of pancreatic adenocarcinomas. Because KRAS itself is a challenging therapeutic target, we focused on understanding key signaling pathways driven by KRAS as a way to reveal dependencies that are amenable to therapeutic intervention. Analyses in primary human pancreatic cancers and model systems revealed that the receptor for the cytokine leukemia inhibitory factor (LIF) is downregulated by mutant KRAS. Furthermore, downregulation of the LIF receptor (LIFR) is necessary for KRAS-mediated neoplastic transformation. We found LIFR exerts inhibitory effects on KRAS-mediated transformation by inhibiting expression of the glucose transporter GLUT1, a key mediator of the enhanced glycolysis found in KRAS-driven malignancies. Decreased LIFR expression leads to increased GLUT1 as well as increases in glycolysis and mitochondrial respiration. The repression of GLUT1 by LIFR is mediated by the transcription factor STAT3, indicating a tumor-suppressive role for STAT3 within cancer cells with mutated KRAS. Finally, reflecting a clinically important tumor-suppressive role of LIFR, decreased LIFR expression correlates with shorter survival in pancreatic cancer patients with mutated KRAS. Similar findings were found in non–small cell lung cancers driven by mutated KRAS, suggesting that silencing LIFR is a generalized mechanism of KRAS-mediated cellular transformation. These results indicate that the LIFR/STAT3 pathway may mediate either tumor-promoting or tumor-suppressive signaling pathways depending on the genetic background of tumor cells, and may play diverse roles within other cells in the tumor microenvironment.
Mutant KRAS drives downregulation of the receptor for LIF, thereby allowing an increase in expression of the glucose transporter GLUT1 and increases in glycolysis and mitochondrial respiration.
Introduction
Pancreatic cancer remains a highly lethal disease. Deaths from pancreatic cancer rank fourth among cancer-related deaths in the United States (1), and the case fatality rate is greater than 90%. One of the hallmarks of pancreatic cancer is aberrant activity of oncogenic KRAS, which is mutated and constitutively activated in 90% of pancreatic adenocarcinomas (2). However, attempts to target KRAS itself have been largely unsuccessful. Understanding downstream events driven by KRAS transformation holds the potential to reveal novel dependencies and therapeutic targets in KRAS-driven cancers.
KRAS-induced transformation elicits a prominent inflammatory response, which is mediated by a range of cytokines, particularly IL6 and its family members, including oncostatin M (OSM) and leukemia inhibitory factor (LIF; ref. 3). This can have a major effect on the tumor cells themselves, through autocrine mechanisms, but also prominent biological effects on cells in the tumor microenvironment including immune, stromal, vascular, and neuronal cells. The principal way in which IL6 and related cytokines regulate gene expression is through the activation of STAT transcription factors, particularly STAT3. IL6 leads to the activation of Jak family kinases associated with the IL-6 receptor, and this leads to the phosphorylation of STAT3 on tyrosine 705. This single phosphorylation event triggers an activating dimerization of STAT3, nuclear translocation, and the binding of STAT3 to nine base pair consensus sites in the regulatory regions of target genes. Although STATs often mediate increased gene expression, they can also lead to the repression of distinct target genes. As such, STAT3 activation can have divergent biological effects, depending on the cellular context.
Because the response of cells to cytokines can be regulated by modulating expression of receptors and downstream signaling proteins, there would seem to be a selective pressure on each cellular component in the tumor microenvironment to allow propagation of prosurvival phenotypes in the setting of chronic exposure to IL6 and its family members. We reasoned that modulation of expression of cytokine receptors in KRAS-transformed pancreatic cancer cells could reveal key aspects of the molecular pathogenesis of this disease, and might suggest novel therapeutic strategies to target cancer cells driven by mutated KRAS.
Materials and Methods
Cell culture, transfection, and establishment of cell lines
AK191 cells were kindly provided by Dr. Ronald Depinho (MD Anderson Cancer Center, Houston, TX) and cultured in DMEM supplemented with 10% FBS. Doxycycline (1 μg/mL) was used to induce mutant KRAS expression in these cells. NIH3T3 cells were obtained from ATCC and cultured in DMEM supplemented with 10% FBS. Mouse mammary epithelial cell line HC11 (from Bernd Groner, Georg Speyer Haus, Frankfurt am Main, Germany) was maintained in DMEM supplemented with 10% FBS. Pancreatic cancer cell lines PANC-1, 8902, CAPAN2, CFPA5, and YAPC were kindly provided by Dr. Andrew Aguirre (DFCI, Boston, MA) and were cultured in DMEM supplemented with 10% FBS.
Mutant KRAS (or empty vector control) was introduced into cells by retroviral infection. Empty vector or human LIFR was introduced into cells by lentiviral infection. To establish inducible STAT3C in the mouse pancreatic cancer cell line AK191, STAT3-C/pGene-stop and pSwitch or pGene-stop and pSwitch were cotransfected into AK191 cells using Lipofectamine 2000 (Invitrogen). Cells were selected in 500 μg hygromycin (Roche Applied Science) and 200 μg zeocin (Invitrogen). Mifepristone (Invitrogen) was used at 0.1 nmol/L for all experiments for induction of STAT3C expression. Cells were maintained in a humidified incubator at 37°C with 5% CO2 and passaged for less than three months after thawing. All cells were authenticated by short tandem repeat DNA profiling, and were routinely tested for the presence of Mycoplasma by PCR.
Reagents
LIF was used at concentration of 10 ng/mL. 5-azacytidine (Sigma) was used at concentration of 1 μmol/L. siRNAs targeting either human KRAS (sc-35731) or GLUT1 (sc-35493) were obtained from Santa Cruz Biotechnology. siRNAs targeting human STAT3 (D-003544-03-0020) or LIFR were from Dharmacon, including siLIFR #9 (J-008017-09), siLIFR #10 (J-008017-10), siLIFR #11 (J-008017-11), and siLIFR #12 (J-008017-12). Nontargeting siRNA control, siControl (D-001210-02), was from Dharmacon. siRNAs were transfected into cells using Lipofectamine RNAiMAX (Invitrogen). A lentiviral vector expressing human LIFR was kindly provided by Dr. Li Ma (MD Anderson Cancer Center, Houston, TX).
Viability and clonogenic assays
Viable cell number was measured by ATP-dependent bioluminescence using the CellTiter-Glo assay (Promega). For clonogenic assays, cells were seeded in 6-well plate at 200–500 cells/well and incubated for 7 to 10 days. Cells were then stained with 6% glutaraldehyde/0.5% crystal violet for 30 minutes, rinsed, and dried.
Immunoblots
Immunoblots were performed using antibodies to phospho-tyrosine 705 STAT3 (9351; Cell Signaling Technology), phospho-serine 727 STAT3 (4), total STAT3 (sc-482; Santa Cruz Biotechnology) phosphor-MAPK (9102S; Cell Signaling Technology), KRAS (14412S; Cell Signaling Technology), LIFR (sc-659; Santa Cruz Biotechnology), actin (A5316; Sigma-Aldrich), FLAG, tubulin (T5168, Sigma-Aldrich).
Quantitative reverse transcription PCR
RNA was harvested using RNeasy Mini Kits from QIAGEN. cDNA was generated using the TaqMan Reverse Transcription kit (Applied Biosystems), and quantitative reverse transcription PCR (RT-PCR) was performed using the indicated primers (Supplementary Table S2). Data are expressed as mean fold change ± SD of three replicates.
TCGA patient survival data analysis
For survival analysis of patients with pancreatic and lung cancer in TCGA, Oncolnc was used for graph production and statistical analysis to compare patients with the highest and lowest third of LIF or LIFR mRNA levels. For subgroup survival analysis of patients with pancreatic and lung adenocarcinoma cancer with KRAS mutation in TCGA, The Cancer Genome Atlas cBioPortal was accessed on December 20, 2016 for overall survival of patients with pancreatic cancers and on January 11, 2017 for overall survival of patients with lung adenocarcinomas. Briefly, LIFR was entered as the query gene. Data were downloaded and manually sorted according to LIFR mRNA level. Data were then entered into Prism for survival curve analysis. Overall survival was compared between patients with the highest and lowest third of LIFR mRNA levels.
Gene-set enrichment and correlation analyses
Gene-set enrichment analysis (GSEA) on TCGA pancreatic cancer dataset, TCGA lung adenocarcinoma dataset and dataset GSE16515 was performed using xapps.gsea.Main from the Broad Institute of MIT and Harvard (http://software.broadinstitute.org; ref. 5) based on Hallmarks gene sets collection (v6.1) from the Broad Institute of MIT and Harvard (http://software.broadinstitute.org). Briefly, data were sorted according to LIFR mRNA level in each individual dataset and each sample was re-named numerically according to LIFR levels. In each dataset, samples with the highest one-third of LIFR mRNA levels were defined as “LIFR-high” phenotype. Samples with the lowest one-third of LIFR mRNA levels were defined as “LIFR-low” phenotype. For gene expression correlation analysis, LIFR and SLC2A (GLUT1) were entered as query genes in cBioPortal (6, 7). The mRNA level for each gene was downloaded and saved in Excel format and subsequently analyzed using Prism-GraphPad. To acquire data related to tyrosine-phosphorylated STAT3, STAT3_PY705 was entered as query gene.
Chromatin immunoprecipitation
Chromatin immunoprecipitation (ChIP) was performed as described previously (8). Briefly, PANC-1 cells were infected with lentiviral vectors with control shRNA or shRNA targeting STAT3. Cells were collected two days after shRNA knocking-down of STAT3. For cytokine-induced STAT3 activation, LIF (10 ng/mL) was added to the cell culture medium 30 minutes prior to harvesting. Cells were fixed in 1% formaldehyde for 10 minutes, sonicated using a Fisher Scientific Sonic Dismembranator Model 500 PDQ, and lysates were immunoprecipitated overnight at 4°C with an antibody for STAT3 (sc-482, Santa Cruz Biotechnology) or IgG control. Quantitative PCR was performed using the indicated primers (Supplementary Table S2), and signal detected was normalized to input.
Quantitation of glycolytic and mitochondrial ATP production
Cells were transfected with siRNA targeting LIFR using siLIFR#9 and siLIFR#10, as well as nontargeting siRNA control as described above. Following transfection, the cells were harvested, counted, and seeded into Agilent Seahorse XF96 cell culture microplates, at previously determined optimal cellular plating densities (2 × 104 cells/well and 1 × 104 cells/well for YAPC and PANC-1 cell lines, respectively). The following day, the cells and calibration cartridge were prepared according to the manufacturer's protocol, by using Seahorse XF Real-Time ATP Rate Assay Kit (Agilent, 103677–100) and Seahorse XFe96 FluxPak (Agilent, 102601–100). Cells were then analyzed for mitochondrial and glycolytic production of ATP on an Agilent Seahorse XFe96 analyzer.
Statistical analysis
For each study, biological or technical repeats were performed as indicated in each figure legend. Results are presented as mean ± SD. Statistical analyses were generated using GraphPad Prism 6 software. Values of P < .05 were considered significant (*, P < .05; **, P < .01; ***, P < .001; ****, P < .0001).
Results
LIFR is downregulated in human pancreatic cancer
IL6 and related cytokines (such as LIF and oncostatin M), which mediate the acute phase response, are often highly expressed in the tumor microenvironment, and plasma levels of these proteins frequently correlate inversely with patient survival (9). Therefore, tumor cells will be selected on the basis of their ability to optimally survive and proliferate in this inflammatory milieu. To understand how the expression of IL6 family cytokines and their receptors are modulated in KRAS-driven cancer, we focused on pancreatic cancer, in which greater than 90% of cases are characterized by activating mutations in KRAS (2). We explored two independent gene expression microarray datasets (GSE16515 and GSE15471) that contain sufficient number of pancreatic cancer samples and normal pancreatic tissues (10, 11). Compared with normal pancreatic tissue, pancreatic cancers express much higher levels of LIF, however, with concomitantly significant lower level of LIFR expression (Fig. 1). In contrast, no difference in IL6 was observed comparing pancreatic cancer with normal tissue. Interestingly, IL6R level and IL6ST (GP130) levels are either similar or higher in pancreatic cancer tissue than in normal tissue, suggesting that the decrease in LIFR expression is not a general response of receptors to this family of cytokines. We also examined expression of three other cytokines, OSM, IL11, and IL27, and their receptors, all of which signal through IL6ST (GP130) as well. All of these cytokines and their receptor show either no difference or they are more highly expressed in pancreatic cancer cells compared to normal tissue. Taken together, these data indicate that in pancreatic cancer LIFR is consistently downregulated, despite an upregulation of the cytokine LIF (Fig. 1).
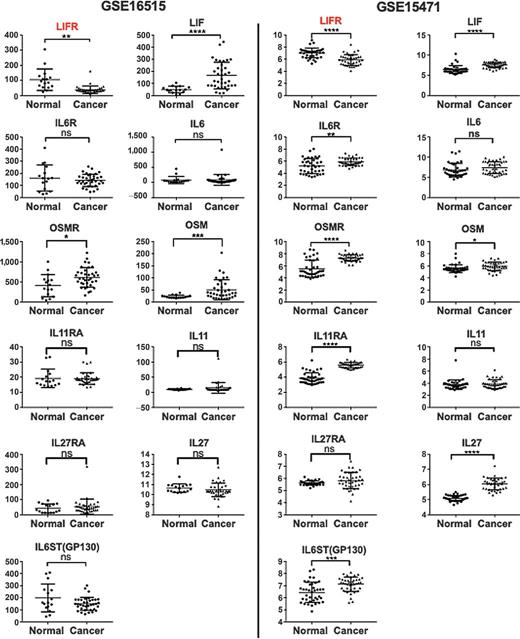
LIFR expression is reduced in human pancreatic cancer. Expression levels of cytokines and their receptors were obtained from gene expression microarray dataset GSE16515 (normal n = 16, cancer n = 36) and GSE15471 (normal n = 39, cancer n = 39). The P value was calculated using an unpaired t test with Welch correction.
LIFR expression is reduced in human pancreatic cancer. Expression levels of cytokines and their receptors were obtained from gene expression microarray dataset GSE16515 (normal n = 16, cancer n = 36) and GSE15471 (normal n = 39, cancer n = 39). The P value was calculated using an unpaired t test with Welch correction.
Oncogenic KRAS inhibits LIFR expression in transformed cells
Given the observation that LIFR is down regulated in human pancreatic cancer, we next considered whether this effect was directly mediated by oncogenic, mutated KRAS, which is found in nearly all of these tumors. We used three isogenic systems, in which nontransformed cells undergo malignant transformation in the presence of mutated KRAS: AK191 pancreatic epithelial cells, NIH3T3 fibroblasts, and HC11 mammary epithelial cells. In each system, we found that expression of mutated KRAS was associated with a prominent decrease in LIFR mRNA expression (Fig. 2A), although KRAS transformation leads to increased expression of LIF (Supplementary Fig. S1A). In AK191 cells, in which expression of mutated KRAS can be regulated by doxycycline, shutting off expression of KRAS is associated with increasing LIFR expression relative to control cells (Fig. 2B). Upon adding back doxycycline, mutated KRAS is reexpressed and LIFR expression rapidly falls to levels consistent with cells harboring persistently activated KRAS (Fig. 2B). To determine the functional significance of the reduced expression of LIFR in KRAS-transformed cells, we added exogenous LIF and measured the phosphorylation of the transcription factor STAT3, a well-characterized mediator of LIF/LIFR signaling. While parental cells showed significant level of STAT3 phosphorylation upon LIF stimulation, KRAS transformed cells showed marked attenuation of STAT3 phosphorylation, consistent with the decreased expression of LIFR on these cells (Fig. 2C).

KRAS transformation suppresses LIFR expression. A, Mutated KRAS was expressed in AK191 cells under control of a doxycycline-responsive promoter. NIH3T3 and HC11 were infected with an empty retroviral vector or a vector expressing mutated KRAS or HRAS, respectively. LIFR mRNA was quantitated by RT-PCR. Protein was prepared from AK191 cells cultured with or without doxycycline for 48 hours. B, AK191 cells were grown in the presence of doxycycline (1 ng/mL) to induce KRAS expression, then had doxycycline withdrawn (time = 0), and readded at 120 hours. RNA was isolated at the indicated times, and LIFR mRNA was quantitated by RT-PCR. C, NIH3T3 cells transduced with an empty vector or NIH3T3 cells transformed with mutant KRAS were stimulated with LIF (10 ng/mL) for the indicated times and protein was isolated for immunoblot analysis (left). AK191 cells were grown in the presence or absence of doxycycline for 48 hours. Protein was isolated after LIF stimulation for 15 minutes (right). D, The indicated pancreatic cancer cell lines were incubated with control siRNA or siRNA to KRAS for 72 hours, after which LIFR mRNA was quantitated by RT-PCR. E, The indicated pancreatic cancer cell lines were incubated with control siRNA or siRNA to KRAS for 72 hours, after which protein was harvested for the indicated immunoblots. F, LIFR protein levels were quantitated (normalized to tubulin) from the immunoblots in E using ImageJ software. Results are mean ± SD of at least three independent experiments. G, LIFR mRNA levels and genomic methylation of this locus in primary pancreatic cancers were retrieved from the TCGA pancreatic cancer provisional datasets and analyzed for correlation. H, AK191 cells were treated with 5-azacytidine (1μmol/L) for 24 or 48 hours, after which mRNA was harvested and analyzed by RT-PCR.
KRAS transformation suppresses LIFR expression. A, Mutated KRAS was expressed in AK191 cells under control of a doxycycline-responsive promoter. NIH3T3 and HC11 were infected with an empty retroviral vector or a vector expressing mutated KRAS or HRAS, respectively. LIFR mRNA was quantitated by RT-PCR. Protein was prepared from AK191 cells cultured with or without doxycycline for 48 hours. B, AK191 cells were grown in the presence of doxycycline (1 ng/mL) to induce KRAS expression, then had doxycycline withdrawn (time = 0), and readded at 120 hours. RNA was isolated at the indicated times, and LIFR mRNA was quantitated by RT-PCR. C, NIH3T3 cells transduced with an empty vector or NIH3T3 cells transformed with mutant KRAS were stimulated with LIF (10 ng/mL) for the indicated times and protein was isolated for immunoblot analysis (left). AK191 cells were grown in the presence or absence of doxycycline for 48 hours. Protein was isolated after LIF stimulation for 15 minutes (right). D, The indicated pancreatic cancer cell lines were incubated with control siRNA or siRNA to KRAS for 72 hours, after which LIFR mRNA was quantitated by RT-PCR. E, The indicated pancreatic cancer cell lines were incubated with control siRNA or siRNA to KRAS for 72 hours, after which protein was harvested for the indicated immunoblots. F, LIFR protein levels were quantitated (normalized to tubulin) from the immunoblots in E using ImageJ software. Results are mean ± SD of at least three independent experiments. G, LIFR mRNA levels and genomic methylation of this locus in primary pancreatic cancers were retrieved from the TCGA pancreatic cancer provisional datasets and analyzed for correlation. H, AK191 cells were treated with 5-azacytidine (1μmol/L) for 24 or 48 hours, after which mRNA was harvested and analyzed by RT-PCR.
This effect of KRAS on LIFR is not generalized to receptors for other LIF family members or related signal transduction proteins. Expression of the IL6 receptor is not reduced in KRAS-transformed cells (Supplementary Fig. S1B). Similarly, no reduction of the shared signal transduction protein IL6ST (gp130) was observed upon KRAS transformation.
Having shown that KRAS expression in nontransformed cells inhibits LIFR expression, we next asked whether mutant KRAS was suppressing LIFR expression in cancer cell lines. We used siRNA to silence expression of KRAS in human pancreatic cancer cell lines, and we observed upregulation of LIFR expression at both mRNA (Fig. 2D) and protein levels (Fig. 2E and F) in all of the lines. Taken together, these data support the hypothesis that KRAS inhibits LIFR expression and that this inhibition occurs at the transcriptional level.
We then investigated the mechanism of LIFR inhibition by KRAS transformation. Because the LIFR gene contains a significant CpG island, we examined whether genomic methylation may play a role in silencing LIFR expression (12, 13). By querying TCGA pancreatic cancer datasets, we found that the level of methylation of the LIFR promoter is inversely correlated with LIFR mRNA expression (Fig. 2G). Furthermore, when pancreatic cancer cell lines were treated with the DNA methylation inhibitor 5-azacytadine, LIFR mRNA expression increased (Fig. 2H). These data indicate that DNA methylation plays a role in silencing LIFR expression in KRAS-transformed pancreatic cancer cells.
LIFR level is associated with overall survival in KRAS-driven cancer
The common finding of silencing of LIFR upon KRAS transformation prompted us to determine whether LIFR affects key cellular pathways and phenotypes in KRAS-driven malignancy. We used RNA interference to knock down LIFR expression in the human pancreatic cancer cell line PANC-1, which possesses high levels of LIFR expression. We observed increased viable cell number and clonogenicity of PANC-1 when LIFR was silenced (Fig. 3A–C). These findings indicate that disruption of LIFR signaling within tumor cells promotes tumor cell survival in vitro.

LIFR inhibits KRAS-driven malignancy. A, PANC-1 cells were incubated with nontargeting control siRNA or siRNAs targeting LIFR, and viable cell number was determined six days after initial transfection. B, Cells were treated with siRNAs as in A, and then protein was harvested three days after transfection. LIFR expression was determined by immunoblotting. C, PANC-1 cells were treated with siRNAs as in A, and after 24 hours, cells were diluted and replated at two different densities. Colonies were stained seven days later. D and E, Survival data were derived from TCGA datasets for pancreatic (D) and lung adenocarcinomas (E) with KRAS mutations. Overall survival is compared between patients in the top and bottom thirds of LIFR mRNA expression.
LIFR inhibits KRAS-driven malignancy. A, PANC-1 cells were incubated with nontargeting control siRNA or siRNAs targeting LIFR, and viable cell number was determined six days after initial transfection. B, Cells were treated with siRNAs as in A, and then protein was harvested three days after transfection. LIFR expression was determined by immunoblotting. C, PANC-1 cells were treated with siRNAs as in A, and after 24 hours, cells were diluted and replated at two different densities. Colonies were stained seven days later. D and E, Survival data were derived from TCGA datasets for pancreatic (D) and lung adenocarcinomas (E) with KRAS mutations. Overall survival is compared between patients in the top and bottom thirds of LIFR mRNA expression.
We next asked whether a similar effect of LIFR also exists in vivo in patients with KRAS-driven cancer. We performed survival analysis comparing patients possessing high LIFR expression in their tumors with patients possessing low LIFR expression in their tumors. We found that a higher LIFR expression level was correlated with better overall survival in pancreatic cancer as a whole (Supplementary Fig. S2A), or in pancreatic cancers with KRAS mutations (Fig. 3D), which represent the vast majority of these cancers. To determine whether this relationship between mutated LIFR expression and survival holds true in other KRAS-driven cancers, we examined lung adenocarcinoma, in which approximately 30% of cases possess KRAS mutation. As with pancreatic cancer, low LIFR expression is associated with shorter survival in lung adenocarcinoma as well (Fig. 3E; Supplementary Fig. S2C). Contrary to the prognostic significance of LIFR, expression of the cytokine LIF is not correlated with overall patient survival (Supplementary Fig. S2B). Furthermore, this relationship is specific for LIFR among receptors for this family of cytokines, as IL6R is not correlated with overall survival in lung adenocarcinoma (Supplementary Fig. S2E). In addition, LIFR level is not correlated with survival in lung squamous cell carcinoma in which only 1.1% of cases possess KRAS mutation (Supplementary Fig. S2D). Thus, LIFR has prognostic significance only in KRAS-driven cancers.
LIFR inhibits glycolysis in KRAS-driven cancer cells
Because LIFR expression affects the phenotype of KRAS-mutant cancer cells and correlates with patient survival, we wanted to take a nonbiased and systematic approach to investigate the mechanism by which LIFR expression affects KRAS-driven neoplastic transformation. We performed GSEA using three publicly available gene-profiling data sets: the TCGA pancreatic cancers with KRAS mutation, TCGA lung adenocarcinoma with KRAS mutation, and the independent pancreatic cancer microarray dataset GSE16515. We first sorted the cases from each dataset according to their LIFR levels. We then performed GSEA comparing the one-third of cases with the lowest LIFR expression with the one-third of cases with the highest LIFR level. We explored whether any hallmark gene sets were particularly enriched in LIFR-low versus LIFR-high cancer. Hallmark gene sets were generated by a computational methodology and represent specific well-defined biological states or processes and display coherent expression (14). From this analysis, we found significantly more cancer hallmark gene sets were enriched in the LIFR-low group compared with the LIFR-high group from all the three datasets (Supplementary Fig. S3). Notably, several metabolic gene sets, including oxidative phosphorylation, reactive oxygen species, hypoxia, adipogenesis, and cholesterol homeostasis, were all enriched in the LIFR-low group (Supplementary Table S1). Importantly, the hallmark gene set “Glycolysis” was significantly enriched in the LIFR-low group from all the three of these data sets (Fig. 4A).

LIFR downregulated glycolytic signatures in KRAS- cancer through modulation of GLUT1. A, GSEA was performed on three different microarray datasets to identify gene sets specifically enriched in LIFR-low group versus LIFR-high group using the 50 Hallmark gene sets. Enriched gene sets were selected on the basis of statistical significance (FDR q value < 0.05 and normalized P < 0.05). B, Correlation analysis was performed for LIFR and SLC2A1 (GLUT1) mRNA levels in The Cancer Genome Atlas (TCGA) provisional datasets for pancreatic cancer and lung adenocarcinoma with KRAS mutation. C, Non-targeting control or siRNAs targeting LIFR were introduced into the indicated human pancreatic cancer cell lines. RNA was isolated 72 hours after transfection and LIFR and GLUT1 mRNA levels were analyzed by RT-PCR. D, Nontargeting control or siRNAs targeting LIFR were reversed transfected into the indicated human pancreatic cancer cells line. Protein was isolated 72 hours after transfection, and analyzed by immunoblotting. E, Human lung adenocarcinoma cell line A549 was infected with an empty vector or a lentiviral vector expressing human LIFR. Cells were then selected in blastomycin for 10 days, after which protein was harvested and analyzed by immunoblotting. F, The indicated cell lines were treated with control or LIFR-targeting siRNA, and ATP production via glycolysis (x-axis) and via mitochondrial respiration (y-axis) was determined. G, Nontargeting control or siRNAs targeting LIFR, KRAS or GLUT1 were introduced into PANC-1 or YAPC cells. Twenty-four hours after transfection, cells were detached, suspended in low-glucose culture medium, and reseeded. Viable cell number was quantitated seven days later by ATP-dependent bioluminescence. Results represent mean± SD of three independent experiments.
LIFR downregulated glycolytic signatures in KRAS- cancer through modulation of GLUT1. A, GSEA was performed on three different microarray datasets to identify gene sets specifically enriched in LIFR-low group versus LIFR-high group using the 50 Hallmark gene sets. Enriched gene sets were selected on the basis of statistical significance (FDR q value < 0.05 and normalized P < 0.05). B, Correlation analysis was performed for LIFR and SLC2A1 (GLUT1) mRNA levels in The Cancer Genome Atlas (TCGA) provisional datasets for pancreatic cancer and lung adenocarcinoma with KRAS mutation. C, Non-targeting control or siRNAs targeting LIFR were introduced into the indicated human pancreatic cancer cell lines. RNA was isolated 72 hours after transfection and LIFR and GLUT1 mRNA levels were analyzed by RT-PCR. D, Nontargeting control or siRNAs targeting LIFR were reversed transfected into the indicated human pancreatic cancer cells line. Protein was isolated 72 hours after transfection, and analyzed by immunoblotting. E, Human lung adenocarcinoma cell line A549 was infected with an empty vector or a lentiviral vector expressing human LIFR. Cells were then selected in blastomycin for 10 days, after which protein was harvested and analyzed by immunoblotting. F, The indicated cell lines were treated with control or LIFR-targeting siRNA, and ATP production via glycolysis (x-axis) and via mitochondrial respiration (y-axis) was determined. G, Nontargeting control or siRNAs targeting LIFR, KRAS or GLUT1 were introduced into PANC-1 or YAPC cells. Twenty-four hours after transfection, cells were detached, suspended in low-glucose culture medium, and reseeded. Viable cell number was quantitated seven days later by ATP-dependent bioluminescence. Results represent mean± SD of three independent experiments.
Glycolysis is one of the key features of KRAS-driven malignancy. Of the 12 human glucose transporter homologs present in the human genome (15), only GLUT1 (glucose transporter-1) is upregulated in mutant KRAS-containing cells compared with wild-type-KRAS-containing cells. Furthermore, enhanced glycolysis in KRAS-driven cancers is dependent on GLUT1 (16). Therefore, we first examined human cancer samples for evidence of a correlation between expression of LIFR and GLUT1 (also known as SLC2A1). We found that in both human pancreatic cancer and human lung adenocarcinoma with KRAS mutation, LIFR expression is inversely correlated with GLUT1 expression (Fig. 4B). To determine whether modulation of LIFR expression directly affects GLUT1 expression, we used RNA interference to deplete LIFR from a panel of human pancreatic cancer cells. This led to an increase in GLUT1 expression both at mRNA (Fig. 4C) and protein level (Fig. 4D). Conversely, overexpression of LIFR led to a decrease in GLUT1 expression (Fig. 4E). We next considered whether changes in LIFR expression directly led to a quantitative change in glycolysis. To do this, we measured both oxygen consumption and extracellular acidification in pancreatic cancer cells in which LIFR expression was decreased with RNA interference. We found that decreasing LIFR expression was associated with a significant increase in both glycolysis and mitochondrial ATP production (Fig. 4F; Supplementary Fig. S4). Taken together, these data indicate that decreased LIFR expression leads to increased GLUT1 expression and increased glycolysis and mitochondrial ATP production in KRAS-transformed cells.
We then asked whether increased GLUT1 expression could account for the increased survival of pancreatic cancer cells upon LIFR silencing. We found that knocking-down GLUT1 at least partially abrogated the increased survival of pancreatic cancer cells upon LIFR silencing (Fig. 4G). These results suggest that LIFR suppresses KRAS-driven malignancy through inhibiting GLUT1 expression.
LIFR activates STAT3 in KRAS-transformed cells
One of the key signaling proteins downstream of LIFR is the transcription factor STAT3. Thus, we considered whether STAT3 was mediating the effects of LIFR in KRAS-driven pancreatic cancer cells. We silenced LIFR in human pancreatic cancer cell lines and observed reduced tyrosine phosphorylation of STAT3 (Fig. 5A) and decreased expression of STAT3 target genes (Fig. 5B). Conversely, overexpressing LIFR increases STAT3 tyrosine phosphorylation in a cell line that has low LIFR expression (Fig. 5C). In addition, the increased expression of LIFR upon KRAS silencing is also associated with increased STAT3 tyrosine phosphorylation (Fig. 2E) and increased STAT3 target gene expression (Fig. 5D). LIFR is coupled through the gp130 signaling chain to JAK family kinases. To determine the family of kinase mediating STAT3 tyrosine phosphorylation, we utilized murine pancreatic epithelial cells, and treated them with a panel of kinase inhibitors. Only a Jak inhibitor abrogated STAT3 tyrosine phosphorylation in these cells (Supplementary Fig. S5), indicating that canonical Jak-STAT signaling was occurring. All of these findings support the contention that LIFR expression correlates closely with, and likely drives STAT3 activation in pancreatic cancer cells.

LIFR promotes STAT3 phosphorylation in KRAS-transformed cells. A, The indicated cells were incubated with nontargeting control siRNA or siRNA targeting LIFR. After 72 hours, protein was harvested for immunoblot analysis. B, The indicated cell lines were incubated with nontargeting control siRNA or siRNA targeting LIFR, after which RNA was harvested for RT-PCR. C, Human lung adenocarcinoma cell line A549 was infected with an empty vector or a lentiviral vector expressing human LIFR. Cells were then selected in blastomycin for 10 days, after which protein was harvested and analyzed by immunoblotting. Data presented are from the same immunoblot as shown in Fig. 4E. D, The indicated cell lines were treated with control siRNA or siRNA to KRAS. After 72 hours (YAPC) or 96 hours (CAPAN2), the expression of STAT3 target genes was quantitated by RT-PCR. Results represent mean± SD of three independent experiments.
LIFR promotes STAT3 phosphorylation in KRAS-transformed cells. A, The indicated cells were incubated with nontargeting control siRNA or siRNA targeting LIFR. After 72 hours, protein was harvested for immunoblot analysis. B, The indicated cell lines were incubated with nontargeting control siRNA or siRNA targeting LIFR, after which RNA was harvested for RT-PCR. C, Human lung adenocarcinoma cell line A549 was infected with an empty vector or a lentiviral vector expressing human LIFR. Cells were then selected in blastomycin for 10 days, after which protein was harvested and analyzed by immunoblotting. Data presented are from the same immunoblot as shown in Fig. 4E. D, The indicated cell lines were treated with control siRNA or siRNA to KRAS. After 72 hours (YAPC) or 96 hours (CAPAN2), the expression of STAT3 target genes was quantitated by RT-PCR. Results represent mean± SD of three independent experiments.
LIFR inhibits GLUT1 through STAT3
Because STAT3 can mediate increased or decreased transcription at distinct genes, we considered the hypothesis that LIFR inhibits GLUT1 through STAT3. We first examined human cancer tissue and found that activation of STAT3 (as assessed by tyrosine phosphorylation of STAT3) is inversely correlated with GLUT1 level in human pancreatic cancer (Fig. 6A). We next determined the effect of STAT3 on GLUT1 expression in cell culture systems. We found that in a number of pancreatic cancer cell lines, silencing STAT3 with RNA interference recapitulates the effect of silencing LIFR in increasing the expression of GLUT1 at both the mRNA (Fig. 6B) and protein levels (Figs. 4D and 6C and D). Similarly, inhibiting STAT3 tyrosine phosphorylation with the JAK inhibitor ruxolitinib upregulates GLUT1 levels (Fig. 6E). Conversely, activating STAT3 with LIF stimulation downregulates GLUT1 levels (Fig. 6F). These results suggest that LIFR inhibits GLUT1 expression through activating STAT3.
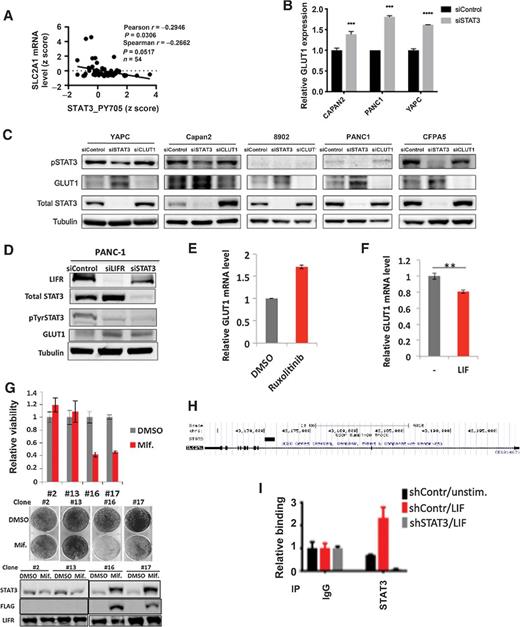
STAT3 inhibits GLUT1 expression in KRAS-transformed cancer cells. A, Correlation analysis was performed between STAT3 tyrosine phosphorylation (STAT3_PY705) and GLUT1 mRNA levels (SLC2A1) from The Cancer Genome Atlas (TCGA) provisional dataset, lung adenocarcinoma with KRAS mutation (n = 54). B, The indicated cell lines were treated with control siRNA or siRNA to STAT3. After 72 hours, mRNA was harvested to quantitate GLUT1 mRNA. C, Pancreatic cancer cell lines were treated with the indicated siRNA for 72 hours (120 hours for YAPC), after which protein was harvested for the indicated immunoblots. D, PANC-1 cells were treated with the indicated siRNA for 72 hours, after which protein was harvested for the indicated immunoblots. E, AK191 cells were treated with the JAK inhibitor ruxolitinib (5 μm) for 6 hours to inhibit STAT3 phosphorylation, after which mRNA was harvested and GLUT1 was quantitated by RT-PCR. F, PANC-1 cells were treated with media or LIF for two hours, after which mRNA was harvested for GLUT1 quantitation. G, Mouse pancreatic cancer cell line AK191 was stably transfected with a mifepristone-inducible constitutively active form of STAT3 (STAT3C) containing a FLAG epitope. Clones with (#16 and #17) and without (#2 and #13) STAT3C responsiveness to mifepristone were isolated, and viable cell number (top) and clonogenicity (middle) was determined in the presence and absence of mifepristone (0.1 nmol/L). Immunoblots were performed to confirm appropriate STAT3C induction (bottom). H, ChIP-seq performed in SKBR3 cells revealed a binding site for STAT3 in the first intron of GLUT1 (black rectangle). I, PANC-1 cells were treated with control shRNA or shRNA to STAT3, and either left untreated or stimulated with LIF for 30 minutes. ChIP was then performed with control IgG or antibody to STAT3 for binding in the first intron of GLUT1.
STAT3 inhibits GLUT1 expression in KRAS-transformed cancer cells. A, Correlation analysis was performed between STAT3 tyrosine phosphorylation (STAT3_PY705) and GLUT1 mRNA levels (SLC2A1) from The Cancer Genome Atlas (TCGA) provisional dataset, lung adenocarcinoma with KRAS mutation (n = 54). B, The indicated cell lines were treated with control siRNA or siRNA to STAT3. After 72 hours, mRNA was harvested to quantitate GLUT1 mRNA. C, Pancreatic cancer cell lines were treated with the indicated siRNA for 72 hours (120 hours for YAPC), after which protein was harvested for the indicated immunoblots. D, PANC-1 cells were treated with the indicated siRNA for 72 hours, after which protein was harvested for the indicated immunoblots. E, AK191 cells were treated with the JAK inhibitor ruxolitinib (5 μm) for 6 hours to inhibit STAT3 phosphorylation, after which mRNA was harvested and GLUT1 was quantitated by RT-PCR. F, PANC-1 cells were treated with media or LIF for two hours, after which mRNA was harvested for GLUT1 quantitation. G, Mouse pancreatic cancer cell line AK191 was stably transfected with a mifepristone-inducible constitutively active form of STAT3 (STAT3C) containing a FLAG epitope. Clones with (#16 and #17) and without (#2 and #13) STAT3C responsiveness to mifepristone were isolated, and viable cell number (top) and clonogenicity (middle) was determined in the presence and absence of mifepristone (0.1 nmol/L). Immunoblots were performed to confirm appropriate STAT3C induction (bottom). H, ChIP-seq performed in SKBR3 cells revealed a binding site for STAT3 in the first intron of GLUT1 (black rectangle). I, PANC-1 cells were treated with control shRNA or shRNA to STAT3, and either left untreated or stimulated with LIF for 30 minutes. ChIP was then performed with control IgG or antibody to STAT3 for binding in the first intron of GLUT1.
These findings suggest that suppression of LIFR expression, with consequential downregulation of STAT3 activation, promotes survival of KRAS-transformed cells. To test this hypothesis, we asked whether forced expression of an activated form of STAT3 would abrogate the protumorigenic effect of KRAS. We employed a cellular system in which an activated form of STAT3 (STAT3C) can be expressed under the control of a mifepristone-inducible promoter (17). We introduced mifepristone-inducible STAT3C into nontransformed AK191 pancreatic epithelial cells. Clones were selected that did (#16 and #17) and did not (#2 and #13) express STAT3C in response to mifepristone. While mifepristone alone had no effects on clonogenesis, overexpressing constitutively activated STAT3 inhibited clonogenesis of KRAS-driven pancreatic cancer cells (Fig. 6G). This finding supports the model that activation of STAT3 counteracts the KRAS-transforming phenotype in pancreatic epithelial cells.
Finally, we considered whether STAT3 could directly bind to the GLUT1 regulatory regions to modulate GLUT1 transcription. We first used data from STAT3 ChIP-seq that we had performed in other cancer systems, and we found that STAT3 can bind to the first intron of GLUT1, a common site of STAT-mediated gene regulation (refs. 8, 18; Fig. 6H). We then performed directed ChIP at this site in PANC-1 cells, and found that STAT3 was able to bind to this site within GLUT1, and that STAT3 binding was further enhanced by LIF stimulation (Fig. 6I). Taken together, these data suggest that STAT3 inhibits GLUT1 transcription by directly binding to this site.
Discussion
Although multiple studies have characterized the array of cytokines produced upon KRAS transformation, the expression of the cognate receptors on KRAS-transformed cells is less well defined. This is an important question, as while these cytokines may have important effects on the tumor microenvironment, relative expression of these receptors on tumor cells can shed light on their effects on tumor biology. We found that, among receptors for the IL6 cytokine family, LIFR is the only receptor that is uniquely and consistently downregulated in pancreatic cancer when compared with normal pancreatic tissue. We then demonstrated that the downregulation of LIFR is mechanistically directly linked to KRAS transformation. This observation is consistent with other reports that showed by immunohistochemistry that LIFR expression was low in pancreatic cancer (19) and LIFR is only expressed on intratumoral nerves in pancreatic cancer tissue (20).
While KRAS transformation inhibits LIFR expression, the cytokine LIF is significantly upregulated in pancreatic cancers. This suggests that while KRAS transformed epithelial cells have a selective advantage in minimizing LIFR pathway activation by downregulating LIFR, increased LIF may contribute to tumorigenesis by modulating other cells in the tumor microenvironment. Indeed, recent work by Bressy and colleagues showed that LIFR is expressed on intratumoral nerves and LIF promotes pancreatic cancer development by driving neural remodeling in pancreatic cancer (20). Although our data indicate that KRAS transformation can directly increase LIF expression in pancreatic cancer cells, it is likely that the majority of LIF production in pancreatic cancer is generated from pancreatic stellate cells, as reported recently (21).
LIFR expression and resultant STAT3 activation clearly antagonize the effects of KRAS-mediated transformation, because knocking down LIFR promotes pancreatic cancer cell survival and activation of STAT3 attenuates the effects of mutated KRAS. These observations are consistent with a recent report by Ma and colleagues (19) who demonstrated that LIFR inhibits metastasis of pancreatic cancer in vitro and in vivo. To determine whether these findings were consistent with data from primary human cancers, we analyzed TCGA survival datasets and found that LIFR expression is positively associated with overall survival in patients with pancreatic cancer. That this finding may be generalizable to KRAS-mediated tumorigenesis is supported by a similar finding in lung adenocarcinomas with KRAS mutation. Importantly, LIFR is not associated with survival in cancers without KRAS mutation, such as lung squamous cells carcinoma. This tumor-suppressive role of LIFR may well be specific to KRAS-transformed cells, as LIFR appears to mediate signaling to promote tumor cell survival in other tumor types or genetic background (22, 23).
Several signaling pathways have been proposed to mediate the tumor suppressive effect of LIFR. Chen and colleagues demonstrated that LIFR inhibited metastasis through the Hippo-YAP pathway in breast cancer (24). Johnson and colleagues found that the LIFR–STAT3 pathway conferred a dormancy phenotype in breast cancer cells disseminated to the bone marrow (25). In pancreatic cancer, it was proposed that LIFR inhibited metastasis by negatively regulating epithelial-to-mesenchymal transition (19). In this study, we used a nonbiased computational approach based on transcriptional signatures to identify pathways modulated by LIFR expression. We found that enhanced glycolysis is consistently enriched in LIFR-low tumor compared to LIFR-high tumor across three independent cancer datasets. Enhanced glycolysis is one of the key hallmarks of KRAS transformation (16). SLC2A1, encoding glucose transporter-1 (GLUT1), is consistently upregulated in cells with mutated KRAS to support enhanced glycolysis. Increased GLUT1 expression and glycolysis is associated with a decreased response to neoadjuvant chemoradiotherapy (26) and gemcitabine resistance in pancreatic cancer (27). We found SLC2A1 expression is inversely correlated with LIFR expression and STAT3 tyrosine phosphorylation in human pancreatic cancer tissue. Furthermore, silencing LIFR in cancer cells upregulates GLUT1 level in KRAS-transformed cells and increases both glycolysis and mitochondrial respiration. These findings reveal a novel pathway by which LIFR exerts a tumor-suppressive role in KRAS-driven cancer.
Our findings also indicate that STAT3 is a key mediator downstream of LIFR, in that it binds to the first intron of GLUT1 to inhibit expression of this gene and the effects of KRAS transformation. This finding adds further weight to the notion that STAT3, although initially described as transcriptional activator, may also exert biologically important effects as a transcriptional inhibitor (28). We observed that in some pancreatic cells, changes in GLUT1 protein level are more substantial than the mRNA level. This could be due to variation of protein/mRNA levels at different time points when cells were collected for analysis. It is unknown whether STAT3 could directly or indirectly modulate the stability of Glut1.
The role of STAT3 in the context of KRAS-mediated transformation remains controversial, with some studies suggesting a facilitating role (3, 29, 30). Furthermore, Shi and colleagues showed that LIF is an important paracrine cytokine in promoting survival and chemotherapy resistance of pancreatic cancer cells (21). However, targeting STAT3 in pancreatic cancer using Jak inhibitors has been unsuccessful (31). On the other hand, several other studies are concordant with our findings, which challenge the traditional purely oncogenic role of STAT3 in cancer driven by mutant KRAS (32–34). These results suggest that STAT3 most likely plays a dynamic, rather than pure oncogenic, role in KRAS-driven cancer. To reconcile these findings, several hypotheses can be considered. First, when phosphorylated on serine727, STAT3 can regulate metabolic function in the mitochondria, which may support Ras-dependent malignant transformation. This function of STAT3 is totally independent of its role as a transcription factor (35). Therefore, in model systems in which STAT3 is genetically ablated, this mitochondrial effect of STAT3 may be dominant, which may obscure the biologically important effect of the transcriptional function of STAT3. Second, it is notable that only LIFR, but not IL6R or other receptors of IL6 family members, is particularly downregulated in KRAS-driven cancer. It is known that a spectrum of transcriptional co-factors may modulate the genomic binding of STAT3 and regulate its transcriptional function (36). It is possible that different cofactors may be activated by LIFR versus IL6R, and thus qualitatively or quantitatively different transcriptional profiles are induced by these cytokines. In addition, it is likely that the LIFR/STAT3 pathway may mediate either tumor-promoting or tumor-suppressive effects, depending on the stage of malignant transformation, differentiation state, and genetic background of pancreatic epithelial cells and it may also play biologically diverse roles among the different cell types within the complex tumor microenvironment. LIF may exert important effects on pancreatic stellate cells, immune cells (37) and neural cells (20) to promote pancreatic cancer cell survival. However, high levels of LIFR expression and STAT3 activation within pancreatic cancer cells themselves counteract cell survival by inhibiting GLUT1 expression and thereby diminishing glycolysis (Fig. 7). This would provide a strong selective pressure to downregulate the LIFR-STAT3 signaling pathway to promote glycolysis and enhance the survival of KRAS-transformed cells in the glucose-deficient microenvironment (16).

Model of the role of the LIF–LIFR–STAT3 signaling axis in tumor progression in KRAS-transformed cells. During inflammation occurring in premalignant pancreatic tissue, LIF is produced by cells in the local environment. LIF acts on many of the cell types in the tissue, including neuronal cells, endothelial cells, stromal cells, and immune cells to promote malignancy. However, within the pancreatic epithelial cells, LIF leads to STAT3 phosphorylation, which represses GLUT1 expression, and limits malignant transformation. With the acquisition of KRAS mutation, LIFR is downregulated, STAT3 phosphorylation is attenuated, and GLUT1 is derepressed, allowing increased glycolysis and malignant transformation.
Model of the role of the LIF–LIFR–STAT3 signaling axis in tumor progression in KRAS-transformed cells. During inflammation occurring in premalignant pancreatic tissue, LIF is produced by cells in the local environment. LIF acts on many of the cell types in the tissue, including neuronal cells, endothelial cells, stromal cells, and immune cells to promote malignancy. However, within the pancreatic epithelial cells, LIF leads to STAT3 phosphorylation, which represses GLUT1 expression, and limits malignant transformation. With the acquisition of KRAS mutation, LIFR is downregulated, STAT3 phosphorylation is attenuated, and GLUT1 is derepressed, allowing increased glycolysis and malignant transformation.
The fact that KRAS-transformed cells express high level of LIF but downregulate LIFR indicates that only a low level of STAT3 transcriptional activity is permissible in KRAS-transformed cells. This reflects the fact that excessive LIF/LIFR/STAT3 signaling, through suppression of GLUT1 and downregulation of glycolysis, will abrogate malignant transformation mediated by mutant KRAS. Thus, the role of STAT3 in KRAS-mediated tumorigenesis is complex and multidimensional.
In conclusion, our findings suggest that downregulation of LIFR expression is necessary for optimal survival of KRAS-transformed pancreatic cancers, as well as other tumors driven by mutated KRAS. This diminishes the tyrosine phosphorylation of STAT3, which derepresses GLUT1 expression. The elucidation of the complex role of the LIFR/STAT3 axis in pancreatic cancer is essential to allow the design of rational therapeutic interventions targeting this pathway (38).
Authors' Disclosures
D.A. Frank reports other support from Brent Leahey Fund and from Betty Ann Blum during the conduct of the study; personal fees from Kymera and Vigeo Therapeutics, grants from NIH, Lustgarten Foundation, Janpix, Biolojic Design, and Cstem, and grants from Roche Genentech outside the submitted work. No disclosures were reported by the other authors.
Authors' Contributions
S. Liu: Conceptualization, investigation, methodology, writing–original draft. H.I. Gandler: Investigation, writing–review and editing. I. Tošić: Writing–review and editing. D.Q. Ye: Investigation, writing–review and editing. Z.T. Giaccone: Investigation, writing–review and editing. D.A. Frank: Conceptualization, supervision, funding acquisition, writing–review and editing.
Acknowledgments
This research was supported by NIH grant R01-CA160979, a grant from the Lustgarten Foundation for Pancreatic Cancer Research, the Brent Leahey Fund, and a generous gift from Ms. Betty Ann Blum (to D.A. Frank).
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.